
Up to 40% of pheos and paras are hereditary.
Other resources:
Watch up-to-date videos on various pheo para genetic topics here.
Watch a presentation on genetics from our 2022 Virtual Pheo Para Conference here.
Find a genetic counselor near you.
Resource from the NIH
Resource from NHS
Genetic Information Nondiscrimination Act
Find out more about how we provide up to date medical information on these pages.
All patients diagnosed with a pheo or para should talk to their doctor about genetic testing. If you have a genetic mutation, your children have a 50/50 chance of inheriting it. Even if your children inherit the gene, that doesn’t mean that they will develop a tumor, but regular monitoring is very important. More information about the pheo para genes and the likelihood of developing a tumor is available below.
Watch this animation on pheo para genetic mutations.
Why is it important?
Recent research indicates that people who know they have a genetic mutation, have less metastatic disease, lower likelihood of complications and better overall health outcomes.
For those with a known genetic mutation, regular (often annual) screening using measurements of plasma-free metanephrines or urinary fractionated metanephrines and catecholamines, together with whole body imaging can catch tumors before they are symptomatic. The frequency and types of screening used as well as the course of treatment is influenced by the specific gene that is altered.
Currently, mutations in over 12 genes are associated with an increased risk of developing pheo para, and geneticists believe more genes will be discovered in the near future. It is important to have genetic testing, not only to identify current mutations associated with pheo para, but also yet to be discovered mutations.
How is it done?
First, you will see a genetic counselor to explain the process and review your family’s medical history. The genetic counselor will determine the correct panel to order based on your personal family history. The actual test is simple: a blood or saliva sample is obtained and sent to a testing laboratory. Next (usually 4-8 weeks later), you will meet with the physician or genetic counselor to discuss your test results. The test can cost between $250-1,000, and is usually covered by insurance, but this is dependent upon your health plan.
Here is what a typical genetic test panel looks like. These panels contain the specific genes that will be tested for genetic mutations. Below is a cancer panel that includes pheo para genes; your panel may only contain pheo para – depending on your personal and family history.

When you receive your results, here are the different categories that your genetic test results can fall under.
Pathogenic mutation (positive) category means that the results almost certainly indicate that you have a genetic mutation, and that your medical team will treat you accordingly, which may include annual testing and/or scanning. Family members may also be offered genetic testing.
Variant, likely pathogenic category means that the results strongly suggest a genetic mutation, and that your medical team will treat you accordingly, likely the same as if you were in the pathogenic mutation category.
Variant, unknown significance (VUS), category indicates insufficient results. This means there is a portion of the gene that looks different from the way it’s normally expected to look. However, researchers haven’t yet confirmed whether this variant is a harmless change or a risk factor to developing pheo para.
If your test results indicate a VUS, it is very important to stay in touch with your genetic counselor or geneticist, because the field of genetics is rapidly changing. As more research is done, there will be new genetic mutations identified and variants will be re-categorized.
Below is a graphic timeline of the discovery of pheo para genes that can be inherited. As you can see, most of the progress has been made recently, and more is expected on the horizon. The graph does not include other somatic genetic mutations that only occur in the cell, not the entire body. Somatic mutations cannot be passed down through families.
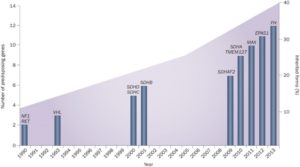
Favier et al. Nature Rev Cancer 2014
Connect with others on Facebook.
Connect with others on Twitter.
Connect with others on Instagram.
Email us at info@pheopara.org to connect one-on-one with another patient.
Join our Community Forum.
I carry a mutation, now what?
If you carry a genetic mutation, regardless of whether you have tumors or symptoms, it is important to be seen by a doctor(s) who has pheo para experience.
If you do not have any tumors or symptoms, regular screening using measurements of plasma-free metanephrines or urinary fractionated metanephrines and catecholamines, together with whole body imaging can catch tumors before they are symptomatic. The frequency of screening is dependent upon your genetic mutation.
You will also need to talk to your family about your status and how they might also be affected. Here is a resource about how to talk to your family. It is standard for genetic counselors to provide you with a letter of explanation that you can share with at-risk family members. It is important to remember that everyone in your family will deal with a possible genetic mutation differently. Some family members will want to take action right away, and other may want to ignore the situation altogether.
Finally, connecting with others who have the same genetic mutation can be comforting, most of these connections happen on social media via facebook pages and in groups. You can also connect with a peer support volunteer through Pheo Para Alliance, and on our community forum at www.pheopara.org.
Here is a list of genes associated with familial pheo para.
NF1, VHL, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, MAX, FH
Some of these genes and the associated syndromes are explained here. Some have only recently been discovered and there is very little research currently available. Some of these genes are also associated with other tumors such as gastrointestinal stromal tumors, renal cell carcinoma and pituitary adenoma, among others.
Which mutations are important?
SDHx Genes
Hereditary Paraganglioma-Pheochromocytoma Syndrome is the result of mutations in the Succinate Dehydrogenase Subunit Genes (SDHx). Patients with mutations in any of the SDH genes are at increased risk for pheochromocytoma and paraganglioma and increased risk of cancerous tumors in the kidney and GI tract. The SDHx genes include four subunits (SDHB, SDHD, SDHC, and SDHA) and an assembly co-factor (SDHAF2).
Here is a table that shows some differences between the SDHx mutations.
| DIFFERENCES BETWEEN SDHx MUTATIONS | |||||
| Genes | Predominant Tumor Site | Multiple/Single Tumors | Family History | Malignancy Risk if tumor develops | Related Conditions* |
| SDHB | para more common than pheo | multiple | low | high | RCC &GIST |
| SDHD | para more common than pheo | multiple | high | low | RCC, GISTS & pituitary adenoma |
| SDHC | paraganglioma | multiple | low | Possibly low | RCC & GISTS |
| SDHA | paraganglioma | single | low | possibly high | GISTS |
| GIST = gastrointestinal stromal tumors | |||||
| RCC = renal cell carcinoma | |||||
| Dahia Nature Reviews Cancer 2014 | |||||
| * Current research indicates that the risk for these tumors is independent. In other words, people who carry an SDHx mutation can develop any of these tumors with pheo paras being the most likely followed by the other related conditions indicated. (Andrews et al Journal of Medical Genetics 2018) | |||||
SDHB
Mutations in SDHB are one of the most common causes of familial pheo/para. Mutations in SDHB are often associated with extra-adrenal pheochromocytomas, parasympathetic paragangliomas, and pheochromocytomas. Pheos and paras that have an SDHB mutation are more likely to be metastatic (cancerous), particularly in younger patients. Someone who carries an SDHB mutation but does not currently have a tumor has a 20-40% chance of developing one by the age of 60 (https://www.ncbi.nlm.nih.gov/books/NBK1548/).
SDHD
Tumors associated with mutations in the SDHD gene are passed down through families by paternal inheritance. If the mutation is inherited from the mother, the children are not at greater risk of developing the disease, but the children still carry the mutation and those children can pass it on to their children. Alternatively, if the mutation is inherited from the father, those children have a greater risk of developing the disease. This can be confusing, for further clarification, please listen to the Liferaft podcast at 33:20. In very rare cases, when SDHD is inherited by the mother, tumors can develop. If symptoms develop, it’s important to consult an experienced medical team.
Patients with SDHD mutations typically present with head and neck paragangliomas but can have paragangliomas anywhere in the body, and these patients are more likely to have multiple tumors. These tumors are usually non-metastatic. Someone who carries a mutation in the SDHD gene passed down by their father, but who does not currently have a tumor, has an approximately 40% chance of developing one by the age of 60 (https://www.ncbi.nlm.nih.gov/books/NBK1548/). The likelihood of metastasis is relatively low, approximately 5%.
SDHC
SDHC is extremely rare, and much research still needs to be done. It is unlikely (but possible) that someone who carries an SDHC mutation but does not currently have a tumor will develop one by the age of 60. The risk for metastasis is relatively low (https://www.ncbi.nlm.nih.gov/books/NBK1548/).
SDHA
SDHA mutations are common in the general population while the development of tumors is rare. In other words, SDHA gene mutations have a low penetrance. This means, that there is only a small chance that someone who carries an SDHA mutation will have a tumor. Current data available suggests that number is as low as 0.1-5% -10% (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6030830/ & https://www.ncbi.nlm.nih.gov/books/NBK1548/). So, you may carry the mutation, but you don’t ever remember anyone in your family having any tumors. Because of this, tumors and SDHA mutations are often found incidentally. In the unlikely event that a pheo para occurs, current research suggests that these tumors may behave aggressively. Because SDHA mutations are relatively common in the population, it is important that individuals known to have an SDHA mutation discuss this with a geneticist during reproductive planning. Babies that inherit two SDHA mutations have a different genetic condition called Leigh syndrome. (https://www.ncbi.nlm.nih.gov/books/NBK1548/)
You can find more information about Leigh syndrome here.
SDHAF2
SDHAF2 is extremely rare, and much research still needs to be done. The risk of developing a tumor from a mutation in this gene (and SDHD) depends upon whether the mutated gene came from the father. This is called paternal inheritance. If the mutation is inherited from the mother, the children are not at greater risk of developing the disease, but the children still carry the mutation and can pass it on to their children. Alternatively, if the mutation is inherited from the father, those children have a greater risk of developing the disease. This can be confusing, for further clarification, please listen to the Liferaft podcast at 33:20.
The tumors associated with SDHAF2 tend to be primarily in the head and neck, and patients with this gene mutations often have multiple tumors. This gene mutation is rare and we do not know the penetrance or likelihood of developing tumors although it is likely low.
VHL
Mutations in the VHL gene lead to Von Hippel-Lindau (VHL) syndrome, which affects approximately 1 in 36,000 people. VHL disease is a genetic condition characterized by tumors in up to ten areas of the body. These tumors can be metastatic.
If an individual has an inherited VHL mutation, each of his or her children will have a 50% chance of inheriting the mutation. Over 90% of patients with this genetic mutation will develop VHL by the age of 65. With careful monitoring, early detection, and appropriate treatment, the most harmful consequences of this gene mutation can be greatly reduced, or in some cases, completely prevented. Approximately 20% of patients with VHL will develop pheochromocytoma. Here are Active Surveillance Guidelines recommended by the VHL Alliance.
VHL syndrome is classified into two types, type 1 and type 2. Patients with VHL type 1 do not develop pheochromocytomas. VHL type 2 is further divided into type 2A (low risk for pheo), type 2B (high risk for pheo), and type 2C (the only symptom is pheo).
More information about VHL can be found here.
NF1
Neurofibromatosis type 1, or von Recklinghausen’s disease, affects approximately 1 in 3,000 people. If an individual has an inherited NF1 mutation, each of his or her children will have a 50% chance of having NF1.
The disorder is characterized by multiple café au lait (light brown) skin spots and neurofibromas (small neoplasms) on or under the skin, and/or freckling in the armpits or groin. About 50% of people with NF1 also have learning challenges. Softening and curving of bones, and curvature of the spine (scoliosis) may also occur. It is rare for a tumor to become metastatic (spread to other parts of the body). NF1 is usually diagnosed in childhood. People who have NF1 are at higher risk of developing pheochromocytoma (approximately 5%-13% life-time risk) compared with the general population.
More information about NF1 can be found here.
MEN2
Multiple endocrine neoplasia type 2 (MEN2) is caused by mutations in the RET gene. It is estimated that about 1 in 30,000 people have MEN2. MEN2 is classified into two subtypes: MEN2A and MEN2B. Both types involve higher risk for development of medullary carcinoma of the thyroid and an increased risk for pheochromocytoma. The lifetime risk for developing a pheo for those who have the RET mutation is approximately 50%.
Patients with MEN2-associated pheo often lack hypertension or other symptoms. If an individual has a RET mutation, then each of his or her children will have a 50% chance of having MEN2. Patients also often have tumors within one or both adrenal glands.
More information about MEN2A and 2B can be found here and here.
Other rare genes associated with pheo para
TMEM127 is a tumor suppressor gene, but its function is not completely understood. It may be involved in transport within the cell. TMEM127 seems to be more commonly associated with adrenal and abdominal pheos. MAX is a transcription factor gene that helps to regulate expression of other genes. Patients with MAX mutations tend to have adrenal pheochromocytomas. Mutations in the FH gene are also associated with pheochromocytoma and paraganglioma. This gene causes a different syndrome called Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC) syndrome. It was discovered that some patients with FH mutations develop pheochromocytoma and paraganglioma. EPAS1 (or HIF2A) mutations are also associated with pheochromocytoma and paraganglioma. These mutations also are associated with polycythemia (high red blood cells or hematocrit) and possibly pancreatic neuroendocrine tumors called somatostatinomas.
Two additional genes, PHD2 and KIF1Bβ, also have been associated with pheochromocytoma/paraganglioma but in extremely rare cases. Other new genes are being identified that may be associated with pheo para but research on them is ongoing.